
Computational Biomolecular Research
We are keen on understanding the mechanisms of enzyme catalysed reactions
employing QM/MM technique. Carbonic anhydrases specifically facinate
us due to their carbon capture properties. To address protein engineering problems,
we use a combination of hybrid electronic structure calculations,
classical force-field simulations and machine learning
to unravel the underlying biophysical-biochemical processes
governing protein stability, dynamics and reactivity.

Computational Catalysis
We use DFT calculations to develop insights into the working
of heterogeneously catalysed reactions. Development of free energy
landscapes of reactions involving carbon capture and utilisation,
clean energy and environment and certain organic reactions are
actively pursued by the research group. Using electronic structure
calculations, we have developed insightes into the working of
the catalysts reported for carbon dioxide hydration,
Suzuki reaction, CO oxidation, formic acid oxidation and
NOx reduction.

Computational Materials Research
Using electronic structure calculations,
we have developed structure-property relationships
governing the catalytic activities of transition metals,
transition and rare-earth oxides and carbonaceous materials.
Our recent focus has been on understanding the behaviour of
transition metal clusters which have been reported to exhibit
exceptional catalytic activities. Our DFT calculations with
catalytic materials enable rational design of
materials. We probe the effects of electronic structure of materials
using state-of-the-art computational methods involving
high-performance computing platforms.
Research @ CMRL

Computational Biomolecular Research
Carbonic anhydrases are ubiquitous in all domains of life and catalyze the interconversion of H2O and CO2 into bicarbonate ions and protons. They are present in all living organisms, encoded as six families viz., α, β, γ, δ, ζ and recently found η class of CAs. Although there is no sequence homology between the families of CAs,
they all contain zinc ions as metal centres in their active sites. Due to the association of CO2 with the reaction they catalyze, they have been reported as potential candidates
in carbon capture, storage and sequestration. Unfortunately, their use in carbon capture is generally limited by their poor stability under reactor operation conditions. Accordingly, there has been an increasing interest in the use of the enzymes extracted from
thermophilic microrganisms (thermophiles) which are stable at high temperatures (323-363
K) and present interesting possibilities as bio-catalyts for carbon capture. Significant attention has
been paid by the scientific community on thermostable enzymes to understand their structural characterstics, origin, mechanism for thermostability and the maximum temperature
limit they can sustain.
Molecular dynamics is a well-established computational tool for studying protein structure and dynamics. This technique has been used to understand and predict structural and functional
details of biological systems at a molecular level. It is capable of providing exhaustive
atomistic information, which may be arduous to obtain by experiments. However, a major
challenge in the implementation of MD is that the associated force-fields are system specific. In biological systems, force-fields can induce bias towards certain types of secondary
structure assignments, which are important for the stability of proteins.
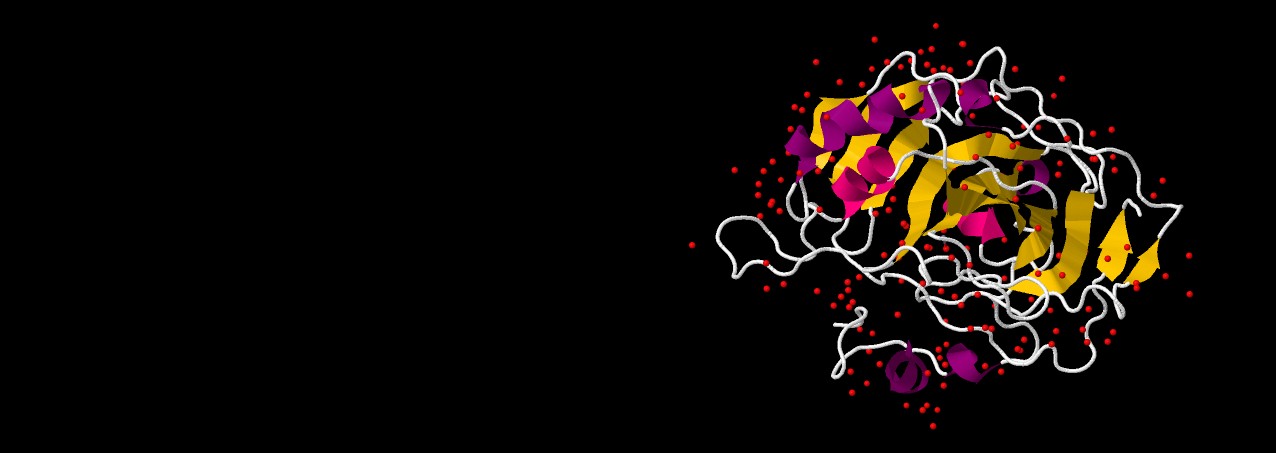
We have explored the force-field dependence of structure
and dynamics of SazCA so as to form a basis for the study of its high temperature behaviour.
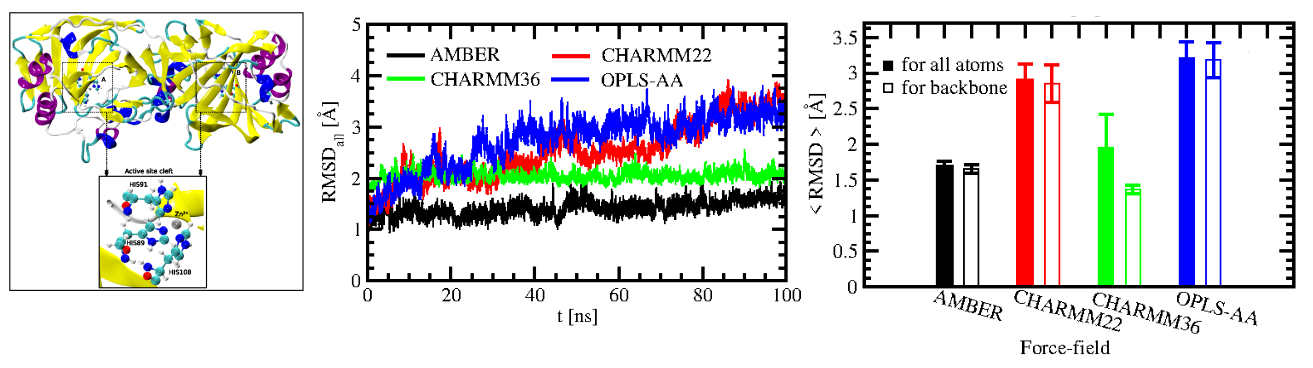 Our room-temperature MD studies on SazCA showed AMBER to be a suitable force field to describe its structure and dynamics [Kumar, Seth and Deshpande, PROTEINS, 2021]
Our room-temperature MD studies on SazCA showed AMBER to be a suitable force field to describe its structure and dynamics [Kumar, Seth and Deshpande, PROTEINS, 2021]
Our high temperature simulations of SazCA, signified by RMSD analysis, suggested that protein was stable upto
353 K. Similar trend was observed with RMSF. The protein maintained its native state upto
353 K, but there was sharp increase in the amplitudes of RMSF beyond 353 K. The amino
acid residues VAL98, ASN99, GLY100, LYS101, GLU145, HIS207 were identified as the
most flexible residues and were responsible for enhanced flexibility of SazCA. The average
fraction of α-helices and β-sheets showed that there was a drastic change in the secondary
structure contents at elevated temperatures which might be the reason for lesser thermostability of SazCA beyond 353 K. Salt bridge analysis showed that ion-pairs (ASP113-LYS81,
ASP115-LYS81, ASP115-LYS114, GLU144-LYS143, and GLU144-LYS206) were responsible
for compromised thermal stability beyond the optimal working temperature. In the nut-shell,
it can be concluded that SazCA maintains its thermal stablity upto its optimal working
temperature and there are structural deformations beyond this temperature majorly due to
a few amino acid residues. Hence, a rational design of thermostable mutant might be
benefited by these observations so as to obtain active, thermostable and industrially viable
carbonic anhydrase for CCUS.
 High temperature MD simulations identified the amino acids associated with compromised thermal stability of SazCA [Kumar, Seth and Deshpande, PROTEINS, 2021]
High temperature MD simulations identified the amino acids associated with compromised thermal stability of SazCA [Kumar, Seth and Deshpande, PROTEINS, 2021]
Biomimetic CO2 Capture Catalysis
Despite its high efficiency and selectivity, carbonic anhydrase has several limitations for direct industrial applications. Since
carbonic anhydrase is soluble in water, reuse of the enzyme is difficult unless immobilized
on a heterogeneous surface compromising its native activity. The
instability of carbonic anhydrase makes it difficult to store it over a long period. Short operational time, cost of extraction after use,
susceptibility to inhibition by small anions and specific operating conditions of pH of 7-10
and temperature of 4-30 deg C make the direct industrial application of carbonic anhydrase impossible. As a general class of catalysts, it is desirable to test the activity of transition metals for biomimetic carbonic anhydrase action. Our DFT calculations with a stable seven atom pentagonal bipyramidal copper cluster showed them to be active as a biomimetic CO2 hydration catalyst exhibiting
carbonic anhydrase-like mechanism. We found the reaction pathway to be identical to that of the α-carbonic anhydrase action involving water adsorption and deprotonation, CO2 interaction and proton transfer, and water attack and bicarbonate
displacement steps. Our calculations in vacuum as well in water as the
solvent indicated insignificant differences in the energetics of the elementary steps in
vacuum and in solvated state indicating the feasibility of the use of Cu as the active species in industrial CO2 capture systems.
 Biomimesis in Cu clusters was indicated by our computations with the mechanism of action same as that of the carbonic anhydrase [Deshpande, Chem Eng Sci, 2016]
Biomimesis in Cu clusters was indicated by our computations with the mechanism of action same as that of the carbonic anhydrase [Deshpande, Chem Eng Sci, 2016]
Inspired from the success of demonstrating Cu clusters as biomimetic, we went ahead and screened a series of transition metals including Ni, Cu, Co, Pd, Ru and Rh for their activities towards CO2 hydration. Using the comparison of energy landscapes developed under the DFT framework, we successfully demonstrated Ni to be a suitable catalyst for liquid phase capture of CO2, as was also experimentally demonstrated by Bhaduri and Siller [Catal Sci Technol, 2013].
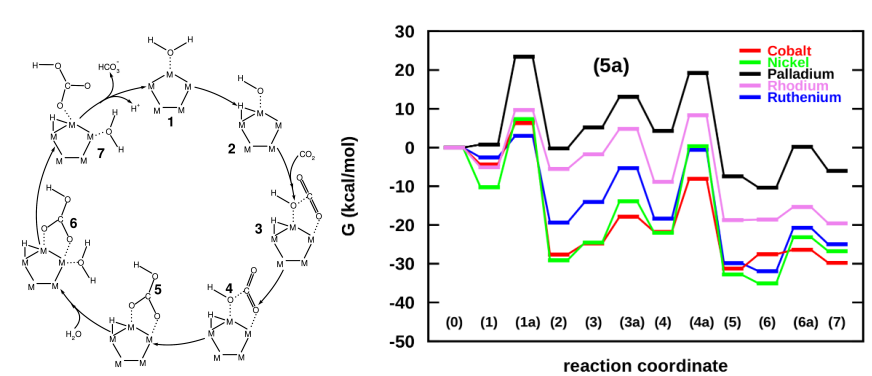 Screening of transition metals for carbonic anhydrase action proved Ni to be the best candidate for biomimetic CO2 hydration [Verma, Sravan Kumar and Deshpande, J Phys Chem C, 2016]
Screening of transition metals for carbonic anhydrase action proved Ni to be the best candidate for biomimetic CO2 hydration [Verma, Sravan Kumar and Deshpande, J Phys Chem C, 2016]
Alkaline saline lakes have significant amounts of different trace salts. Borate ions have been reported to absorb CO2 on the surfaces of water bodies and
CO2 capture by water bodies is attributed to borate catalyzed CO2 hydration. Guo et al. [Env Sci Technol, 2015]
conducted detailed experimental studies on borate-catalyzed CO2 hydration and proposed that
CO2 hydration catalyzed by borate ions followed the carbonic anhydrase mechanism. With an aim of developing insights into the mechanism of CO2 hydration with borate species, molecular details of the elementary steps of CO2 hydration over borate-based catalysts for biomimetic CO2 hydration were developed by us under the DFT framework. Our study established a correspondence between experimental and computational observations and provided rationale for further development of borate-based catalytic systems for carbon capture with potential applications to flue gas treatment. We proved that borate, triborate and tetraborate ions followed the reaction mechanism of CO2 hydration reaction
catalyzed by carbonic anhydrases. Hydroxyl centres in the borate compounds acted as the active sites for CO2 hydration. The elementary steps of the reaction involved parallel CO2 complex
formation which was endergonic, bent CO2 complex formation which was an activated process, bicarbonate ion formation, and bicarbonate displacement by water molecule. Bicarbonate ion formation
followed the Lindskog mechanism involving bicarbonate ion formation following internal rotation.
This pathway was found to preferable over the internal proton transfer with smaller activation barriers.
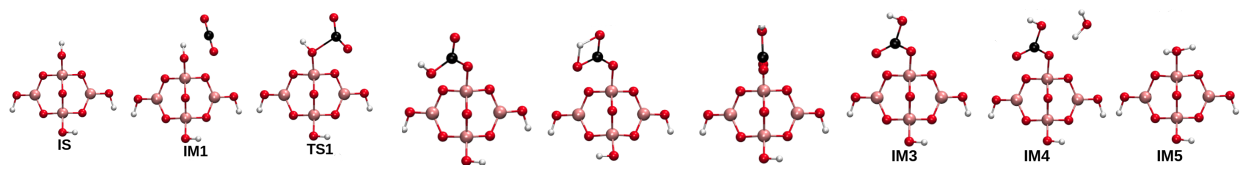 Elementary steps of CO2 hydration over borate, triborate and tetraborte ions proved the catalysts to be biomimetic [Verma and Deshpande, J Phys Chem C, 2017]
Elementary steps of CO2 hydration over borate, triborate and tetraborte ions proved the catalysts to be biomimetic [Verma and Deshpande, J Phys Chem C, 2017]
Structure-property Relationships and Catalysis by
Reducible Oxides
Reducible oxides like CeO2 and TiO2 are established redox catalysts with vital applications in clean energy and environment. Reducibility of these oxide is the key to their successful functioning and efforts have been made by investigators across the globe to enhance their reducibilities. In line with this idea, we have been engaged in developing structure-property relationships in transition metal substituted CeO2 and TiO2 and their effects on catalysis using DFT calculations. We have proposed the formation of differential bond lengths in the lattice of solid solutions to be responsible for the improvement in their catalytic activity. Our DFT analysis of ceria-based solid solutions (Ce1–xMxO2−δ (M = Zr/Mo/Pd)) identified the role of cation substitution and vacancy formation in bond length distribution. A large improvement in the net oxygen activation was observed with the introduction of a cation in the lattice with feasible vacancy formation close to the substituent. Our study successfully explained the reported high activity of Ce1–xPdxO2−δ for redox reactions following dual site mechanism allowing the interaction of surface species in close proximity.
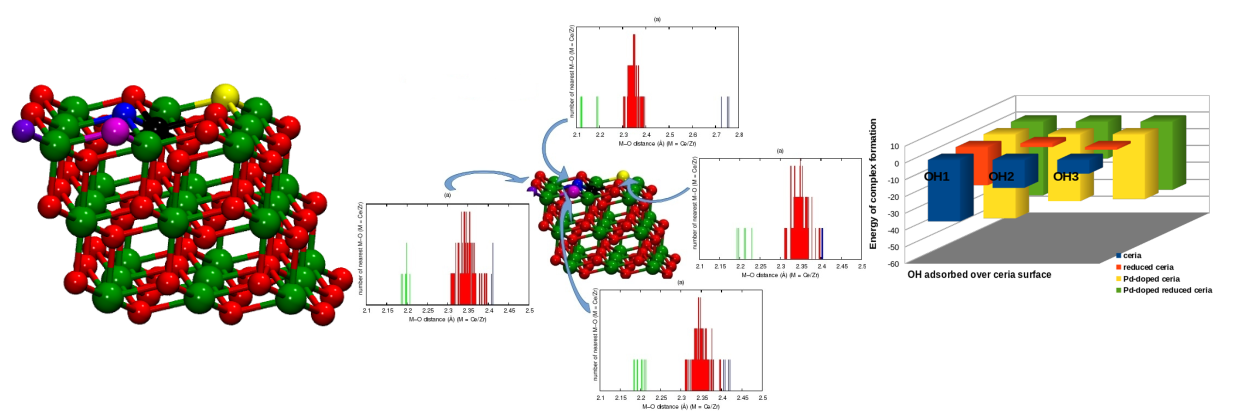 Bond length distribution quantifying the net oxygen activation and identifying the best site for vacancy in Ce1–xMxO2−δ (M = Zr/Mo/Pd) [Sravan Kumar and Deshpande, J Phys Chem C, 2015]
Bond length distribution quantifying the net oxygen activation and identifying the best site for vacancy in Ce1–xMxO2−δ (M = Zr/Mo/Pd) [Sravan Kumar and Deshpande, J Phys Chem C, 2015]
Utilizing these observations, we probed the elementary surface processes duing CO oxidation over Ce1–xPdxO2−δ. Mechanism of CO oxidation over ceria-based catalysts has been widely accepted to follow Mars–van Krevelen (MvK) mechanism involving the lattice oxygen exchange. Adsorption of CO over the metallic sites, lattice oxygen exchange, and replenishment of the support by stream oxygen are considered to be the surface processes taking place during the MvK mechanism. We revealed an alternative MvK-type mechanism for the reaction taking place over metal ion substituted ceria-based reduced catalysts involving hopping of vacancies. The mechanism was proposed on the basis of adsorption energetics of CO and O2, probed with the help of DFT calculations. Our computational analysis showed the adsorption of O2 to take place over the reduced catalyst and nonsequential adsorption of CO and O2 to take place over Ce1–xPdxO2−δ in contrast to sequential adsorption of O2 followed by CO over CeO2−δ. We conclusively highlighted the differences in the elementary surface processes in the proposed mechanism with the established MvK mechanism with the major observation being the differences in the lattice oxygen abstraction and vacancy hopping observed with newly proposed alternative MvK mechanism.
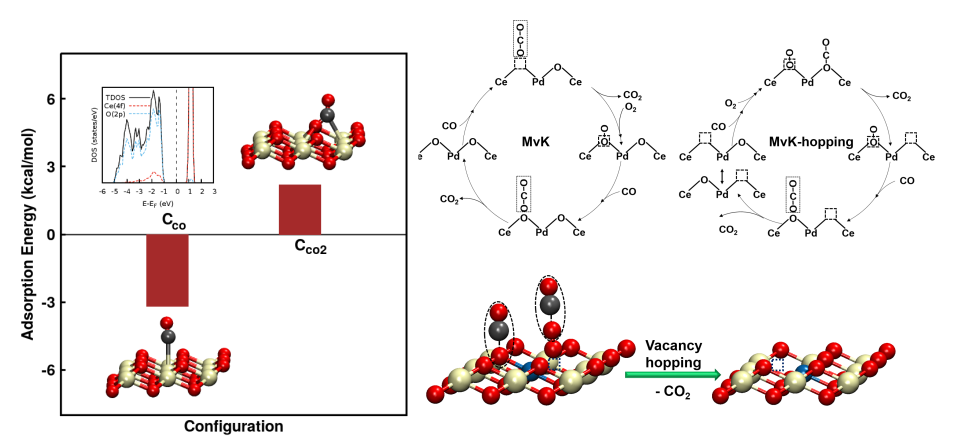 A mechanism alternative to the conventional MvK mechanism involving vacancy hopping releaved using DFT [Pentyala and Deshpande, Ind Eng Chem Res, 2019]
A mechanism alternative to the conventional MvK mechanism involving vacancy hopping releaved using DFT [Pentyala and Deshpande, Ind Eng Chem Res, 2019]
CeO2-based catalysts due to the
presence of alternate Lewis acid-Lewis base sites have a potential of catalyzing C-C coupling
reactions in which oxidative addition is an important step. We analysed adsorption and oxidative addition of halobenzenes over CeO2. Our DFT calculations of a periodic slab of CeO2 (111) with cationic and
anionic surface defects revealed the role of surface defects of both types on adsorption of halobenzenes. We showed the synergistic effect of presence of cationic substitution and anionic vacancies which contributed to an increase in Lewis acid-base interactions resulting in improved
adsorption characteristics. On the basis of these fundamental observations, we demonstrated successfully the working of CeO2-based catalysts for Suzuki-Miyaura reaction. The same concepts were extended to rational design of catalyst for acetylene hydrogenation.
 Acid-base interactions on the surface of Pd-substituted CeO2 probed using DFT. Different electronic effects in O-plane and Ce-plane demostrated the role of each species in the catalytic activity. [Padole and Deshpande, J Phys Chem C, 2016; Padole et al., Phys Chem Chem Phys, 2017]
Acid-base interactions on the surface of Pd-substituted CeO2 probed using DFT. Different electronic effects in O-plane and Ce-plane demostrated the role of each species in the catalytic activity. [Padole and Deshpande, J Phys Chem C, 2016; Padole et al., Phys Chem Chem Phys, 2017]
Crystal planes govern the activities of catalytic materials with TiO2-based materials being no exceptions. Our group has carried out a series of DFT-based studies to probe the dependence of exposed crystal plane on the activities of substituted titania. We have dedicated our attention to develop structure-property relationships in TiO2-based solid solutions with particular reference to net oxygen activation. Density functional theory calculations were implemented on (001) and (100) planes of anatase TiO2 with anionic vacancy defects, cationic substitution (Ti1−xPdxO2) and solid solutions with anionic vacancy defects (Ti1−xPdxO2−δ). (100) plane was found to show higher oxygen activation with smaller rigidity towards surface relaxations. Vacancy formation energy was found to decrease with an introduction of cationic defects. Different vacancy sites on the two planes were observed to show different oxygen activations. Formation of surface hydroxyl groups, envisaged as dissociative adsorption of H2 over oxidized catalyst or dissociative adsorption of H2O over reduced catalyst, was found to be influenced by cation substitution with the stability of surface hydroxyls being compromized by substitution.
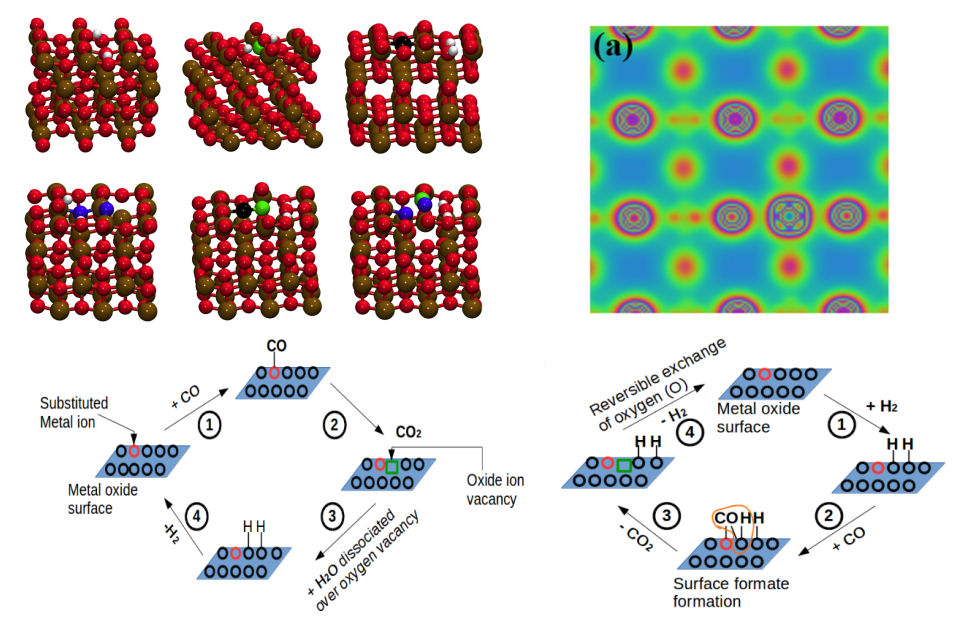 Identifying suitable chemical enviroment in TiO2 solutions to improve catalytic properties [Phani Kumar and Deshpande, Phys Chem Chem Phys, 2017, 2020; Comput Mater Sci, 2017, 2018, 2019]
Identifying suitable chemical enviroment in TiO2 solutions to improve catalytic properties [Phani Kumar and Deshpande, Phys Chem Chem Phys, 2017, 2020; Comput Mater Sci, 2017, 2018, 2019]
Structure-property Relationships and Catalysis by
Heterofullerenes
Chemically modified fullerenes are emerging as an important class of technological materials. For rational design of such advanced materials, structure-property relationships need to be probed. This can help us identify various applications of the newly synthesized materials and also for synthesizing novel fullerene-derivatives with the cage tailored to behave in a desired manner. Our group has focused on heterofullerenes and developed insights into the structure-property relationships in such compounds. Our DFT calculations have established the role of foreign atoms in tailoring the electronic structure of this class of materials.
 DFT calculations identified the role of foreign atoms on the structure and properties of heterofullerenes [Padole and Deshpande, Mol Sim, 2019]
DFT calculations identified the role of foreign atoms on the structure and properties of heterofullerenes [Padole and Deshpande, Mol Sim, 2019]
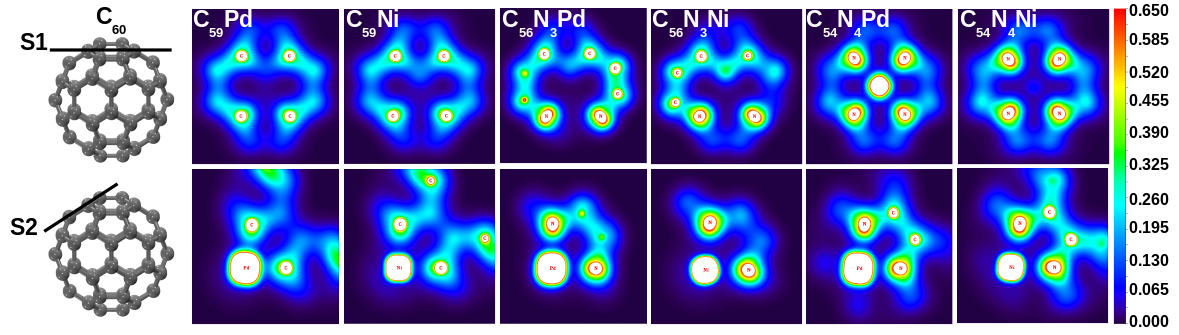
Appreciating the importance of transition metals in catalysis, we tested the catalytic properties of heterofullerenes for various reactions. We carried out DFT investigations of palladium and nickel substituted fullerenes probing the changes in their surface adsorption potential. Structure and
bonding in these heterofullerenes were established with insights into metal-carbon bond
character, stability and adsorption potential. We used C2-gases as probe gases for adsorption
tests with adsorption of acetylene, ethylene and ethane studied with different sites over pure
and heterofullerenes. We established surface modification of the fullerene molecule with a foreign metal to be responsible for enhanced
the gas-substrate interactions. Enhanced surface interactions and differential
adsorption behaviour of different heterofullerenes made them potential candidates as selective
acetylene hydrogenation catalysts. Free energy landscapes for hydrogenation of acetylene and
ethylene over all three compounds were developed. The energy barriers for various elementary
steps during hydrogenation were significantly smaller over the heterofullerenes when compared to
those over C60.
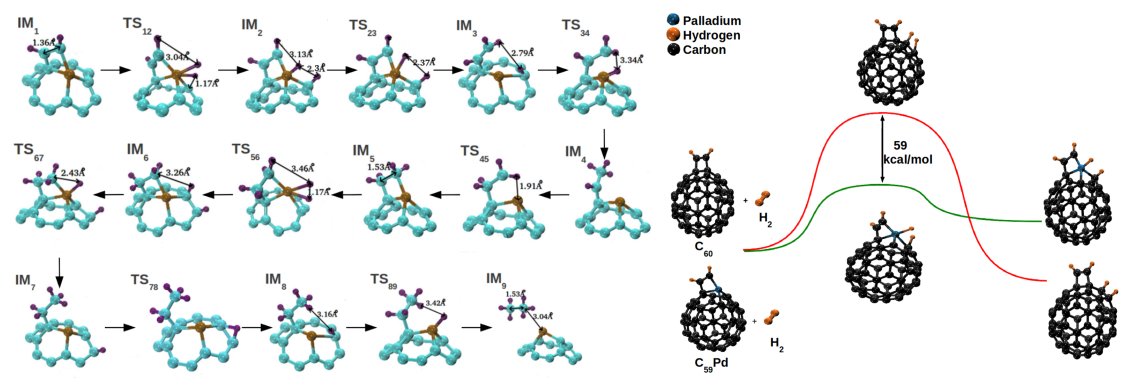 Our DFT calculations established Pd and Ni substituted fullerenes to be potential C2 hydrogenation catalysts with Pd being superior for achieving ethylene selectivity [Padole and Deshpande, J Phys Chem C, 2016]
Our DFT calculations established Pd and Ni substituted fullerenes to be potential C2 hydrogenation catalysts with Pd being superior for achieving ethylene selectivity [Padole and Deshpande, J Phys Chem C, 2016]
We extended this analysis of catalytic properties of heterofullerenes to Suzuki-Miyaura and Heck reaction. Potential of Pd and Ni-substituted fullerenes for oxidative addition of halobenzenes was
investigated using DFT. The metal centers in the catalysts were found to be
the potential reaction sites. We found the adsorption of halobenzenes to be mildly exergonic over both the
catalysts. Activation of all halobenzenes was observed over both the compounds. Oxidative
addition of iodobenzene was the least energy intensive process with a free energy
requirement which was three times smaller than that of fluorobenzene. Activities of both the
catalysts were found to be comparable. We also developed complete free energy landscapes of Suzuki-Miyaura and Heck reaction over both the catalysts providing the details of elementary steps of the reaction.
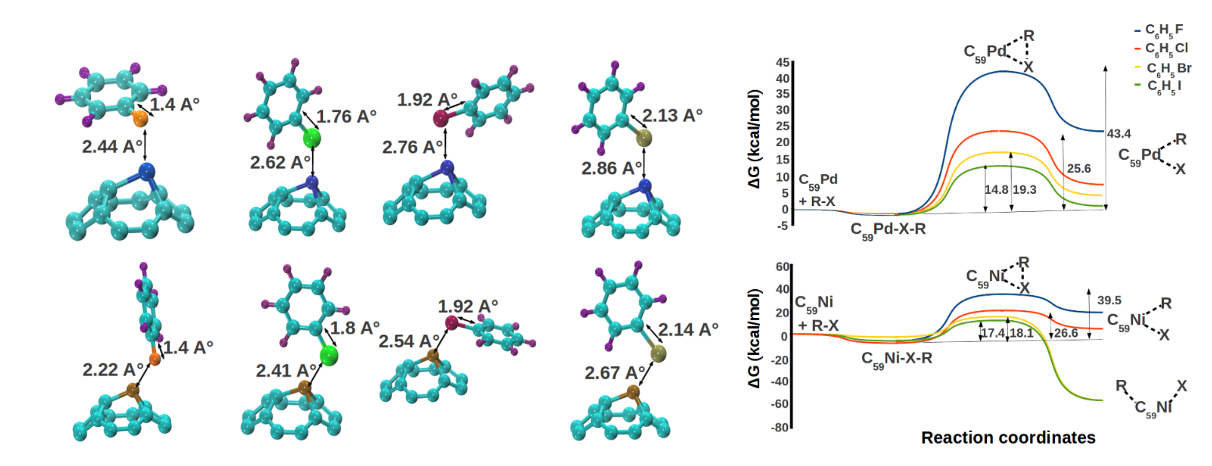 The potential of Pd and Ni substituted fullerenes was revealed by DFT calculations for catalysing Suzuki-Miyaura and Heck reactions [Padole and Deshpande, J Phys Org Chem, 2017]
The potential of Pd and Ni substituted fullerenes was revealed by DFT calculations for catalysing Suzuki-Miyaura and Heck reactions [Padole and Deshpande, J Phys Org Chem, 2017]