Research
- Light Harvesting Complex in Plant Thylakoid Membrane
- Lipid Polymorphism in Plant Thylakoid Membrane
- Membrane Response to Guest Molecules
- Thermodynamic Origin and Stability of Asymmetric Bilayers
- Coupling between Water and Membrane Dynamics
- Dual scale Simulations of Surfactant, Co-surfactant, Water systems
- Dynamics of Water near Soft Interfaces
- Interaction of Protein with Biomolecules
- Molecular Dynamics Simulations of Fenugreek-Inflammasome Adaptor Protein Complex:
-
Multiscale simulation of light harvesting complex
- Molecular Dynamics Simulations of Gel to Liquid Crystalline Transition of Bilayer
-
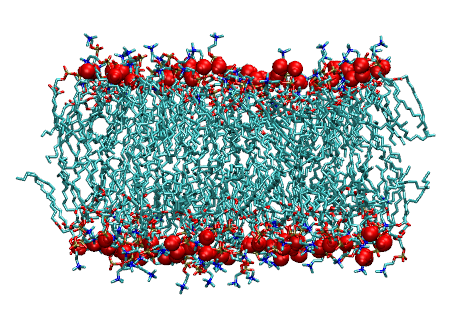
Dynamics and Thermodynamics of Hydration layers of Lipid Bilayer
We compute the entropy and transport properties of water in the hydration layers of DPPC bilayer by using a recently developed theoretical scheme based on the velocity autocorrelation function (VACF) and the power spectrum. Water near the bilayer head groups have substantially lower entropy than the bulk water molecules. The translational diffusion constant for the first hydration layer of water is an order of magnitude smaller than that of bulk water molecule confirming the bound nature of water near lipid head groups. The behavior of translational mean square displacement is going to normal diffusive to subdiffusive region from bulk to surface water indicating caging effect near surface. We calculate rotational diffusion constant from the slope of reorientational mean square displacement of dipole and bond vector of water. We find that rotational diffusion constant near surface is lower than that of bulk which is supported by the slower reorientational relaxation of dipole vector near surface than that of bulk confirming bound nature of water near surface. The two relaxation time constants for reorientation associated with legendre polynomial of order 1 and 2 indicate the presence of angular jump with the diffusion dynamics. -
Dynamics and Thermodynamics of Hydration layer of Bilayer across phase transition
-
Kink mechanism for barrier crossing by long chain molecules
Barrier crossing of polymer is a widely studied problem due to the nature of the underlying physics and its strong relevance to biology. We consider the Kramers problem for a long chain polymer trapped in a biased double well potential. Initially, the polymer is in the less stable well and it can escape from this to the other well by the motion of its N beads across the barrier to attain the configuration having lower free energy. In one dimension we simulate the crossing and show that the results are in agreement with the kink mechanism suggested earlier. In three dimensions, it has not been possible to get analytical kink solution for an arbitrary potential. However, one can assume the form of the solution of the non-linear equation as a kink solution, and then find a double well potential in three dimensions. To verify the kink mechanism,
simulations of the dynamics of a discrete Rouse polymer model in a double well, in three dim ensions were done. We find that the time of crossing is proportional to the chain length in agreement with the kink mechanism. The shape of the kink solution is also in agreement with the analytical solution, in both one and three dimensions. -
Polymer in a double well: dynamics of translocation of short chains over a barrier
-
Dynamics of translocation of star Polymer in a double well
-
Rate Processes with Dynamical Disorder - A direct Variational Approach
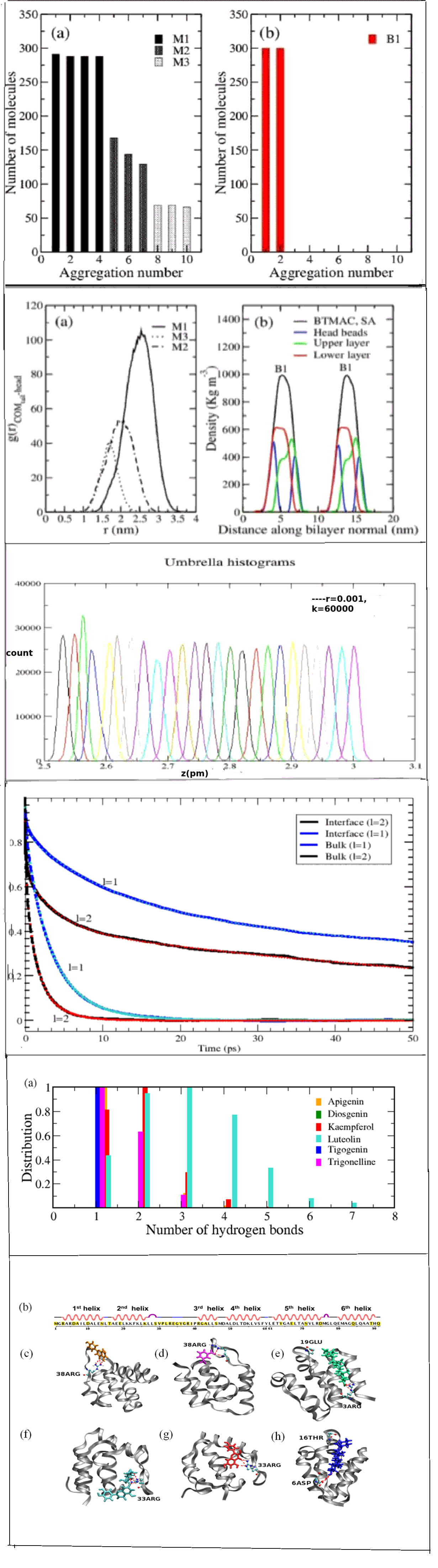
The light harvesting complex (LHCII), pigmented protein trimer, from plants captures energy from sunlight and transfers to the photosynthetic reaction center during photosynthesis. Chlorophyll (chl) molecules are indispensable pigment molecules bound to the light harvesting complex II (LHCII) which is a pigment binding trimeric protein with specific binding sites. They act like antenna during photosynthesis to capture light and transfer to the photosynthetic reaction center in green plants. The photosynthetic pigments of higher plants exist in complex oligomeric states. The interaction of chl and other porphyrinoids with membranes has received extensive attention recently due to several reasons. Chl molecules work in collective fashion in photosynthetic reaction for which their cooperative behavior is important to study. Their interactions with membranes can be used in the important area of photodynamic therapy for an anticancer treatment. Chls through strong absorption, allow light penetration into tissues when associated with proper carriers and high phototoxicity to tumor cells with no toxicity to healthy ones. All these biological properties are cooperative in nature and dependent on how they are assembled. The role of association of pigments and lipids on the stability of the LHCII remains elusive till date. We develop and use coarse-grained models of pigments in plant thylakoid membranes in the absence of the LHCII to characterize and explain experimentally observed phenomenon such as fluorescence quenching. We focus on understanding the role of lipidome on the stability of the protein and the cofactors affecting the architecture and structural dynamics of thylakoid using multiscale simulations.
------------------------------------------------------------------------------------------
Thylakoid membranes provide the cell boundary of chloroplast in plants. They provide the matrix to the photosynthetic light harvesting complex which captures energy from sunlight and transfers to the photosynthetic reaction center. The membrane comprises of different kinds of lipids among which close to 50% is non-bilayer lipids. Due to the presence of the non-bilayer lipids the thylakoid membrane is known to undergo lipid polymorphism leading to phase transitions from the lamellar to non-lamellar phases, such as inverted hexagonal phase or co-existence of inverted micellar phase and lamellar phase. However, the driving force of the phase transition and the molecular insights to its modulation by the protein complex are not clear. The close association between the non-bilayer lipids and the protein complex also appears to be important for the higher-order, multilamellar, onion-like structures adopted by the thylakoid, which are somewhat similar to those in the grana. Thus the effect of the non-bilayer lipids on the membrane topology and the effect of the cross-talk between the lipid, the protein and the cofactor on the protein function are enigmatic aspects of membrane biology, particularly in photosynthesis.
------------------------------------------------------------------------------------------
Understanding the response of membranes towards guest molecules is crucial in various biological processes and have significant implications in fields such as drug delivery, cell banking, artifical photosynthetic materials. Our research focuses on how guest molecules, such as drugs, pigments and cryoprotectants, alter membrane properties. We examine the potential of bilayer as carriers for drugs, emphasizing their ability to encapsulate and deliver drugs at targeted sites. In addition, we investigate the influence of cryoprotectants on membranes at phyiological temperature, which is crucial for understanding the permeation behavior and fluidity of membranes. Leveraging multiscale modeling, we focus on the structural and dynamic changes caused by guest molecules on membranes while elucidating the fundamental thermodynamic principles. The response of lipids to guest molecules can be pivotal in better designing of chemical approaches to drug delivery, cryopreservation, artificial light harvesting with improved efficacy and safety.
------------------------------------------------------------------------------------------
Core Research Grant, Science and Engineering Research Board, DST
(Year: 2020-23)
The naturally existing bilayers are mostly asymmetric in nature and act as a selective barrier for the passage of extracellular fluids or ions within the cell. The difference in compositions across two leaflets results in asymmetric inhomogeneous bilayers. These are vital for a number of metabolic phenomena such as thrombosis, phagocytois etc. We aim to understand the origin and stability of asymmetry in biological membranes using multi-scale modeling.

------------------------------------------------------------------------------------------
Water is the most abundant molecule in cells. Water molecules near membranes affect several biological processes such as transport of drugs and small molecules across the cell, influences formation of membrane rafts, molecular recognition, signal transduction and so on. In the past decade, with a major advancement of computer simulations and experimental techniques, water near bio and soft interfaces are found to have distinct properties with slow relaxations compared to that of the bulk water and these water molecules are termed as biological water. However, dynamics of biological water from membrane experiments remain fragmentary due to the fluidity of membranes at physiological temperature and inaccessible atomistic trajectories. Moreover, the influence of water on global dynamics of membrane or protein are still debated. The hydration water dynamics near lipid membranes are investigated using all atom molecular dynamics simulations. Chemically confined interfacial water and lipid membranes exhibit distinct relaxation rates manifesting dynamical heterogeneity. We aim to find the correlation between relaxation time scales of interface water and hydrogen bonded lipid moieties so that hydration dynamics can act as a sensitive reflector of regional membrane dynamics.
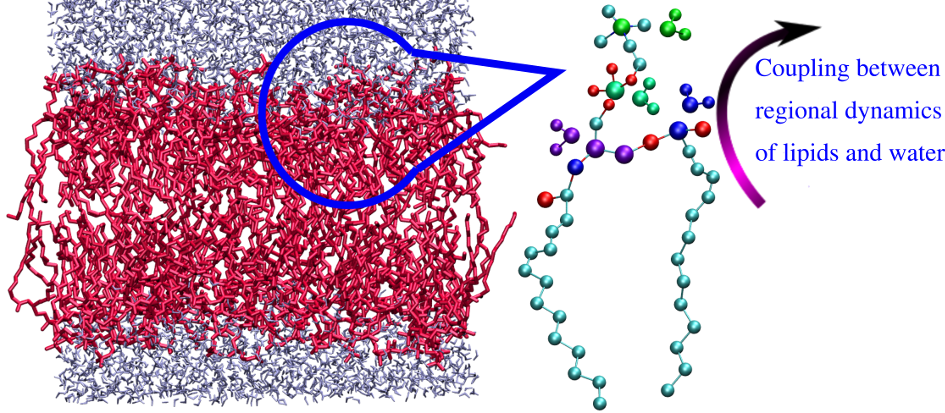
------------------------------------------------------------------------------------------
Fast track Young Scientist Scheme, Science and Engineering Research Board, DST
(Year: 2014-17)
Since surfactants have immense industrial applications in cosmetic formulations and oil recovery processes, structural and thermodynamical properties of these complex fluids are fundamentally important to design phases according to the desired functionalities. Understanding the inter-relationships between various parameters of surfactants which play crucial roles in dictating the stability of different micro-structures remains a challenge till date. This work focuses on understanding the influence of composition and temperature on the formation of topologically distinct phases of mixed surfactant co-surfactant bilayers using multi-scale modeling techniques. The study of phase modulations with composition has implications on other biophysical properties such as fusion and pore formation.
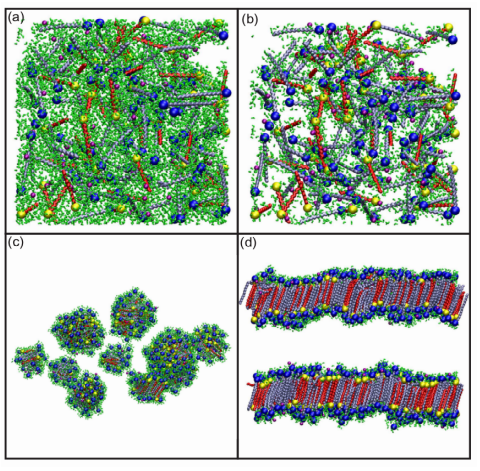
------------------------------------------------------------------------------------------
In this work we investigate the dynamics of hydrogen bond networks near interfaces and bulk to understand how molecular jump mechanism plays a role in the phase change of a bilayer.
---------------------------------------------------------------------------------------
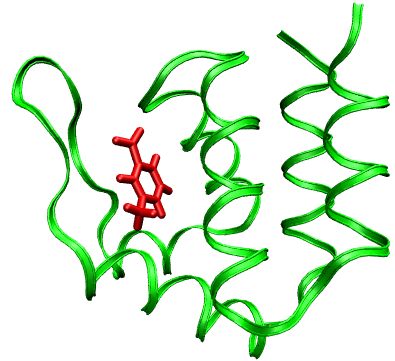
The majority of human kidney stones are composed primarily of calcium oxalate monohydrate.(COM) crystals. Thus, determining the molecular modulation of COM crystallization by urinary constituents is crucial for understanding and controlling renal stone disease.Interactions between proteins and crystals are thought to be of great importance in many, if not all, forms of biomineral-ization. Such interactions have been proposed to be responsible for the nucleation of crystals in mineralizing tissues, the inhibition of crystallization in soft tissues formation of particular crystalline polymorphs,and biological control over crystal orientation,growth habit.The aim is to obtain the general insights of peptide-crystal interaction.

In collaboration with Ateeque Malani.
---------------------------------------------------------------------------------------------------
Trigonella foenum graecum (fenugreek) has been known as an important plant in traditional Indian medicine for its anti-inflammatory properties for long time. Although fenugreek is widely accepted as an anti-inflammatory agent with cytoprotective effects, the molecular mechanism is still not clear. Using molecular simulations, we show that the protein can bind to individual phytochemicals of fenugreek via specific non-covalent interactions without altering the protein conformations.
-------------------------------------------------------------------------------------------
Previous Work
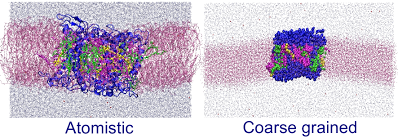
Light harvesting complex is a trimeric protein pigment complex which collects energy from sunlight and transfers to the photosynthetic reaction center in plants.The structural properties of the light harvesting complex ( LHCII) are investigated to understand the trimerization process of the protein monomers. With all atom molecular dynamics simulation we study various specific interactions focusing on the role of protein and pigments on trimer formation and stabilty. Based on the atomistic level simulations we derive a CG model for LHCII with a particular attention to chlorophyll (CHL) b/a, a pigment in LHCII. The structural properties and partitioning behavior of CHL in the lipid bilayer are consistent with the higher resolution description. The dynamical properties of CHL b/a in membrane are also investigated in different levels of resolutions. Using the CG model we aim to study the large scale aggregation of protein trimer for the current biomolecular system.
------------------------------------------------------------------------------------------
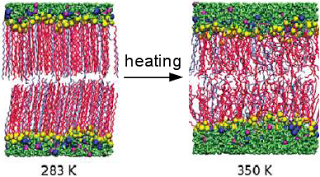
Bilayers are important constituents of membranes having two layers of surfactant or lipid molecules. Lipid bilayers have important roles in living organism whereas the bilayer comprising surfactant or co-surfactant has immense application in detergents, cosmetic formulations, and secondary and tertiary oil recovery processes. Depending on the ratio of the projected hydrophobic core volume to the projected area of the surfactant, water−surfactant systems can arrange into a variety of structures ranging from micelles, bilayers, reverse micelles, and vesicles. There are a large number of experimental studies available in literature which investigate the structural properties and phase behavior of these systems, but does not give insights in the molecular levels. In that respect, molecular dynamics simulation is an attractive tool which can provide microscopic three dimensional structural information. The gel to liquid crystalline transitions in the bilayer by varying the bilayer components is studied. The bilayer is found to exist in tilted phase at low temperatures and for the compositions investigated in this study. The tilted to L$_\alpha$ melting transition occurred in the same temperature range as obtained in the experiment. The larger area per head group associated with the quarternary amine surfactant decreases the transition temperature with an increase in the surfactant composition. For the highest surfactant composition, we observe an increase in the d-spacing prior to the melting transition, accompanied by a sharpening in the water density variation across the head group region of the bilayer. Signatures of this swelling effect are observed in the alkane density distributions, area per head group and membrane thickness and attributed to the hydrophobic effect. At a fixed bilayer composition, the tilted to L$_\alpha$ transition temperature obtained for the high water content bilayer is similar to that obtained with low water content, confirming that the melting transition at these water contents, is dominated by chain melting.
------------------------------------------------------------------------------------------
------------------------------------------------------------------------------------------
Hydration layer of biomolecules are of great importance in recent research because of its major role in determining the structures, stability and dynamics of biomolecules such as reverse micelles, vesicles and membranes. Water molecules in the hydration layer are the key parameters that modulate the biomolecular functionalities such as intercalation, membrane stability, fusion, enzyme catalysis, protein and molecular recognition. Dynamical and thermodynamical properties of water in the hydration layers of bilayer are calculated to study the effect of bilayer melting on the water dynamics across transition. The translational diffusion constant follows arrhenius behavior till bilayer melting transition from gel to liquid crystalline state and there is a dynamic crossover after the melting transition. The area per head group increases across the melting allowing more available space for associated water molecules near the head group having higher translational and rotational degrees of freedom. This is corrobated with sharp increment of entropy and free energy across the transition. This proposes a new method to predict the melting temperature of bilayer from water dynamics.
------------------------------------------------------------------------------------------
------------------------------------------------------------------------------------------
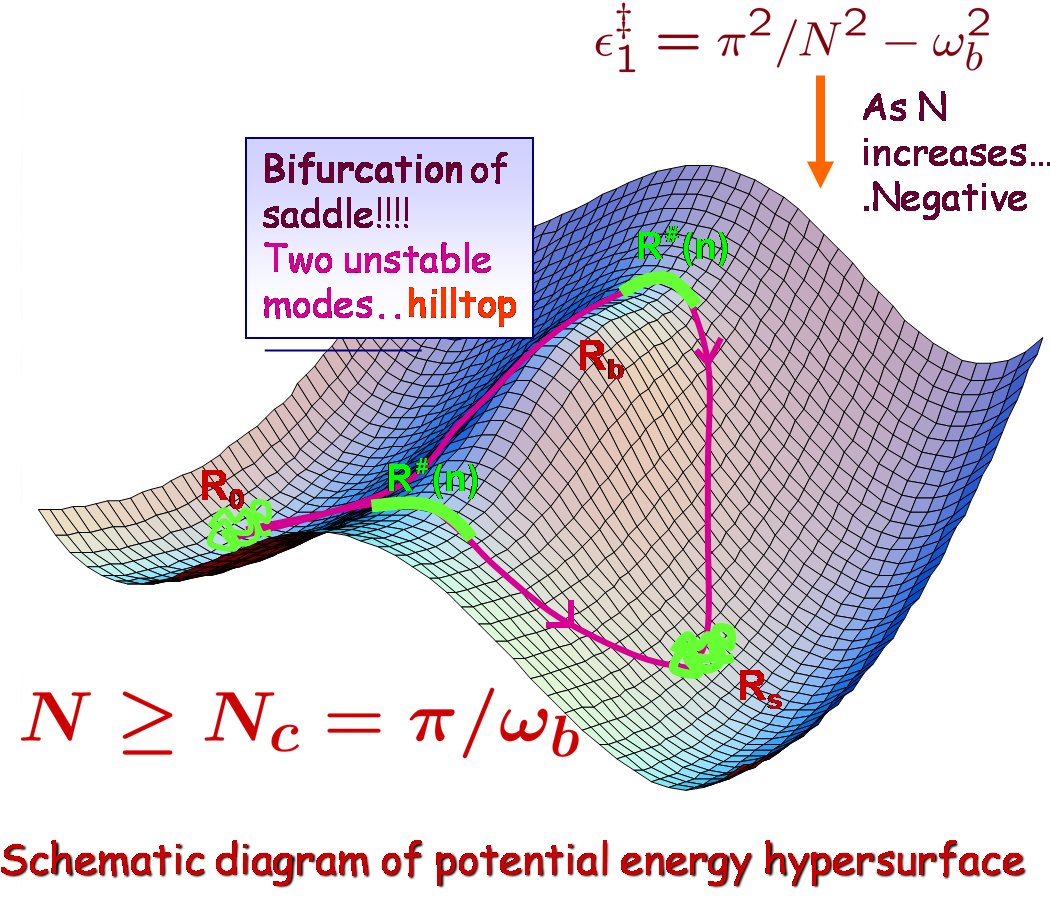
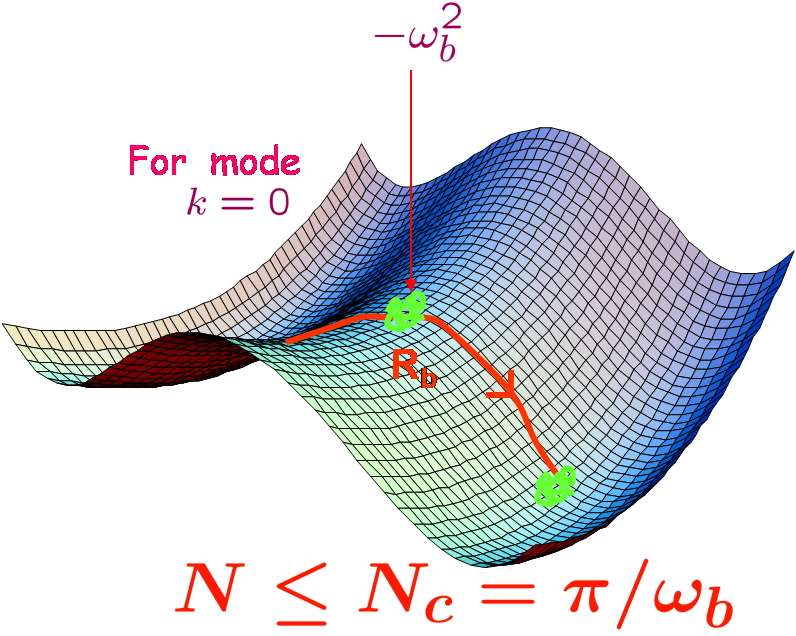
We consider the dynamics of a short chain polymer crossing over a free energy barrier. We find exact expression for the activation energy and rate of crossing using continuum version of Rouse model. To solve the transition state of polymer one has to solve Newton-like equations of motion for a fictitious particle. The analysis shows that if the chain length is below a critical length, chain crosses the barrier as a globule, and if it is above the critical length it has an extended structure. When it is a globule, the activation energy would increase linearly with the polymer length. But stretching out lowers the energy and hence the activation energy is no longer linear in length. The rates in both the cases are calculated using multidimensional approach and a new formula is derived which avoids the infinite product calculation with the help of Coleman's approach. However, the harmonic approximation in the derivation makes the rate diverging at the point where the saddle point changes over from globule to stretched conformation where bifurcation of saddle to two new saddles occurs. A correction formula is derived for the rate in the vicinity of the point usinghigher order expansion in potential. Using those analytical expressions we get rate as a function of length numerically.
------------------------------------------------------------------------------------------
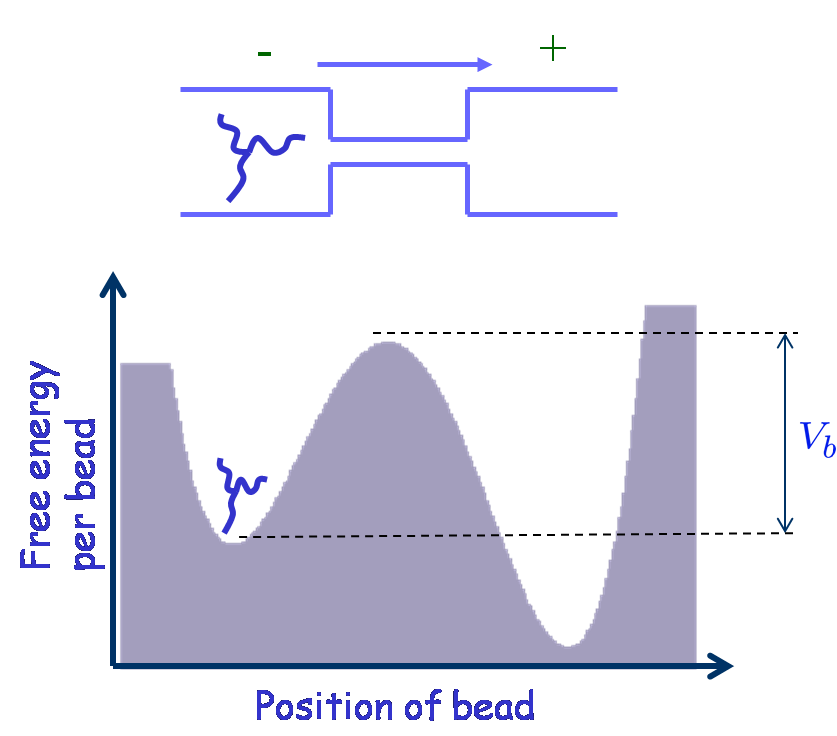
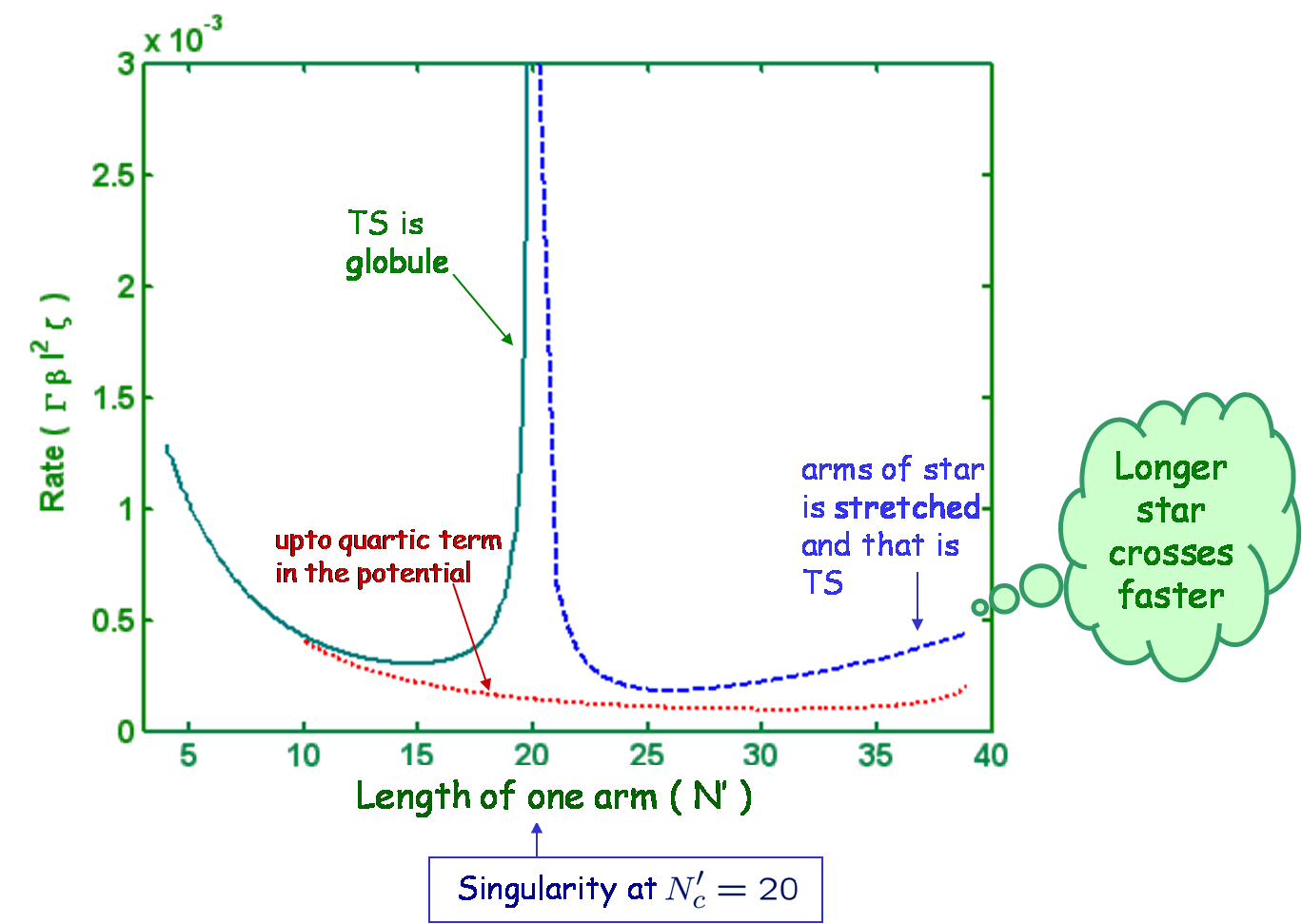
We analyze the dynamics of a star polymer of F arms confined to a double well potential. Initially the molecule is confined to one of the metastable wells and can cross over the barrier to the other side. We use the continuum version of Rouse-Ham model and calculate the rate of crossing using the multidimensional approach due to langer (Ann. Phys. 54, 258 (1969)). For each star polymer, there is a critical total length, N_{Tc}, below which the polymer crosses over as a globule. In such a case the activation energy is proportional to the total arm length of the star. Above N_{Tc} the star crosses the barrier in a stretched state. Thus, there is a multifurcation of the transition state at N_{Tc}. Above N_{Tc}, the activation energy at first increases and then decreases as one increases the arm length. We calculate the rate by expanding the energy around the saddle up to second order in the fluctuations which has a prefactor of infinite products. We show that these infinite products can be reduced to a simple expression, and evaluated easily. However, the rate diverges near N_{Tc} due to the multifurcation, which results in more than one unstable mode. To get rid of this divergence we keep terms upto fourth order in the expansion of energy for these modes. Performing this, we have calculated the rate as a function of the length of the star. It is found that the rate has a nonmonotonic dependence on the length, suggesting that longer stars may actually cross over faster.
------------------------------------------------------------------------------------------
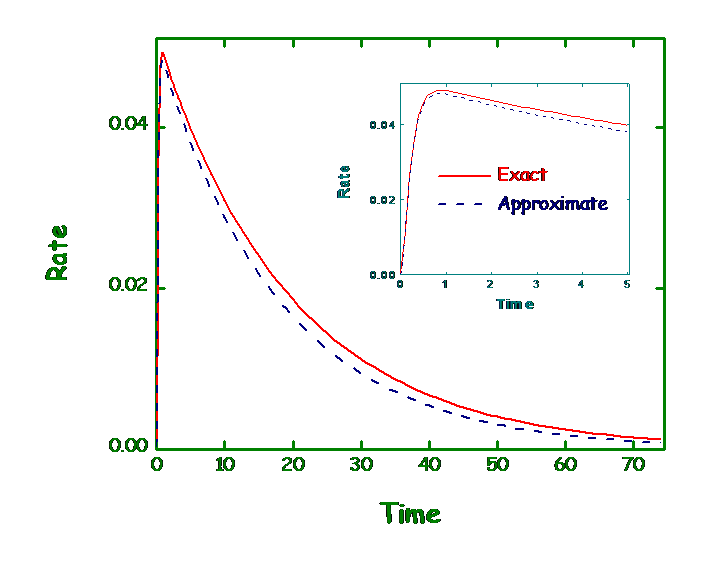
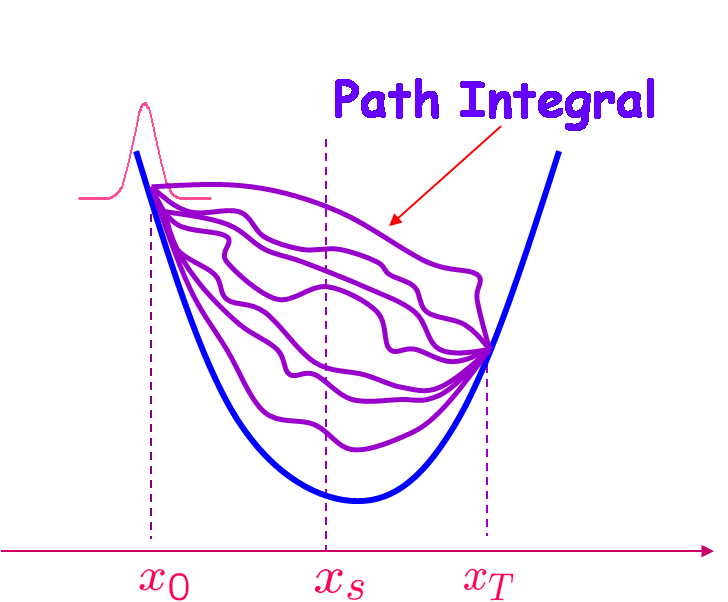
We develop variational approximations using path integral approach to the survival probability for rate processes with dynamical disorder. The exact solution of corresponding Smoluchowski equation was found earlier analytically in laplace domain with sink having arbitrary strength and position. Zwanzig has proposed an indirect approach to the calculation of survival probability involving chemical reactions occurring in fluctuating medium. Variational approximation to this calculation was done in laplace domain also. In real domain direct exact analytical calculation is not possible. So we use the direct approximate variational path integral technique to calculate both lower and upper bound of survival probability in time domain. We mimic the delta function sink by quadratic sink for which the path integral can be solved exactly. The strength of the quadratic sink is treated as variational parameter and using the optimized value for it, one can estimate the optimized lower as well as upper bound of survival probability. We also compare the variational results with the exact numerical calculation.
-----------------------------------------------------------------------------------------------